




有源醫療器械加速老化試驗效期驗證及使用期限驗證流程
為了確保醫療器械在生命周期內的安全及有效性,注冊人應在設計開發中對其使用期限予以驗證。使用期限的驗證是醫療器械可靠性研究的重要組成部分,制定并驗證合理的使用期限有


注冊備案 · 臨床試驗 · 體系建立輔導 · 分類界定 · 申請創新
Recommend case

來源:醫療器械注冊代辦 發布日期:2019-10-29 閱讀量:次
引言:第二批試點醫療器械注冊人省份單位中,僅剩遼寧省暫未發布實施方案或征求意見稿。為滿足山東省內各醫療器械企業需求,山東省藥監局在結合國家藥監局要求和省內醫療器械企業情況,制定了符合山東省現實情況的試點工作實施方案。
")
魯藥監注〔2019〕58號
山東省藥品監督管理局關于印發《山東省醫療器械注冊人制度試點工作實施方案》的通知
各市市場監督管理局,省局相關處室、單位:
《山東省醫療器械注冊人制度試點工作實施方案》已經局務會審議通過,現印發給你們,請遵照執行。
山東省藥品監督管理局
2019年10月28日
(公開屬性:主動公開)
按照國家藥品監督管理局《關于擴大醫療器械注冊人制度試點工作的通知》(國藥監械注〔2019〕33號)(以下簡稱《通知》)要求,結合我省實際,制定本實施方案。
通過開展醫療器械注冊人制度試點工作,改革完善醫療器械注冊審評審批和生產制度,落實醫療器械注冊人的主體責任,落實跨區域監管責任。優化醫療器械資源配置,激發創新研發活力,推動醫療器械創新發展,加快臨床急需醫療器械上市,更好滿足公眾高質量用械需求。
(一)探索建立醫療器械委托生產管理制度,優化資源配置,落實主體責任。醫療器械注冊申請人(以下簡稱“申請人”)申請并取得醫療器械注冊證的,成為醫療器械注冊人(以下簡稱“注冊人”)。
申請人可以委托《通知》試點區域內具備相應生產能力的企業生產樣品并申請醫療器械注冊證。注冊人具備相應生產資質和能力的可以自行生產,也可以委托試點區域內的生產企業生產產品;受托生產企業不具備相應生產資質的,可提交注冊人的醫療器械注冊證申請生產許可辦理。鼓勵集團公司通過注冊人制度試點進一步整合、優化資源配置,落實醫療器械注冊人主體責任。
(二)探索建立完善的注冊人醫療器械質量管理體系,明確醫療器械注冊人、受托人等主體之間的法律關系,在責任清晰、風險可控的基礎上,構建注冊人全生命周期質量管理制度和體系。
(三)探索創新醫療器械監管方式,有效落實“監管工作一定要跟上”的要求,完善事中事后監管體系,厘清跨區域監管責任,形成完善的跨區域協同監管機制,增強監管合力,提升監管效能。
(四)探索釋放醫療器械注冊人制度紅利,鼓勵醫療器械創新,推動醫療器械產業高質量發展。
(五)積累醫療器械注冊人制度試點經驗,為全面推進實施醫療器械注冊人管理制度提供重要支撐。
1.本方案委托生產醫療器械范圍包括境內第二類、第三類醫療器械(含創新醫療器械)。
2.屬于原國家食品藥品監督管理總局發布的禁止委托生產醫療器械目錄的,暫不列入本方案的委托生產品種范圍。
(一)注冊人條件
1.住所或者生產地址位于山東省內的企業、科研機構。
2.具備專職的法規事務、質量管理、上市后事務等工作相關的技術與管理人員,具有醫療器械監管法規和標準相關知識和經驗。
3.建立與產品相適應的質量管理體系并保持有效運行,有對質量管理體系獨立進行評估、審核和監督的人員。
4.具備承擔醫療器械質量安全責任的能力。
(二)注冊人的義務責任
1.依法承擔醫療器械設計開發、臨床試驗、生產制造、銷售配送、售后服務、產品召回、不良事件報告等環節中的相應法律責任。
2.與受托生產企業簽訂委托合同和質量協議,明確委托生產中技術要求、質量保證、責任劃分、放行要求等責任,明確生產放行要求和產品上市放行方式。
3.加強對受托生產企業的監督管理,對受托生產企業的質量管理能力開展質量評審,定期對受托生產企業開展質量管理體系評估和審核。
4.加強不良事件監測,根據風險等級建立醫療器械相應的追溯管理制度,確保醫療器械產品可滿足全程追溯的要求。
5.可以自行銷售醫療器械,也可以委托具有相關資質的醫療器械經營企業銷售。自行銷售的注冊人應當具備規定的醫療器械經營能力和條件,無需辦理醫療器械經營許可或者備案;委托銷售的,應當對受委托銷售的醫療器械質量負責,并加強對受托方經營行為的管理,保證其按照法定要求進行銷售,應當與受托方簽訂委托合同,明確各方權利、義務和責任。
6.通過信息化手段,對研發、生產、銷售和不良事件監測情況進行全流程追溯、監控。
7.確保提交的研究資料和臨床試驗數據真實可靠、系統完整、可追溯。
8.發現受托人的生產條件發生變化,不再符合《醫療器械生產質量管理規范》要求的,應當立即要求并監督受托人采取整改措施確保符合《醫療器械生產質量管理規范》的要求。可能影響醫療器械安全、有效的,應當立即要求受托人停止生產活動,及時召回存在缺陷的醫療器械,并向所在地省級藥品監督管理部門報告。
(一)受托生產企業條件
1.住所或者生產地址位于參與試點的省、自治區和直轄市內的企業。
2.具備與受托生產醫療器械相適應的質量管理體系和生產能力。
(二)受托生產企業義務責任
1.承擔《醫療器械監督管理條例》、其他相關法律法規以及委托合同、質量協議規定的義務,并承擔相應的法律責任。
2.按照醫療器械相關法規規定以及委托合同、質量協議約定的要求組織生產,對注冊人負相應質量責任。
3.發現上市后醫療器械發生重大質量事故的,應當及時報告所在地省級藥品監管部門。
4.受托生產終止時,受托生產企業應當向所在地省級藥品監管部門申請減少醫療器械生產許可所附生產產品登記表中登載的受托產品信息。
5.受托生產企業不得再次轉托。
6.受醫療器械注冊人委托代為銷售時,應當具備相應的醫療器械經營條件,符合相關法規要求,辦理醫療器械經營許可或者備案。
(一)注冊申請。申請人按照試點工作要求,委托本省或試點省(市)具備相應醫療器械生產能力的企業生產樣品并申報第二類醫療器械注冊的,由山東省藥品監督管理局(以下簡稱省局)根據注冊申報資料、委托合同和質量協議等有關證明注冊人條件和受委托生產企業條件的材料,組織開展注冊質量管理體系核查(跨省委托的則組織跨省延伸檢查)。經審查符合要求的,核發醫療器械注冊證,核發醫療器械注冊證中登載的生產地址為受托生產地址,備注欄標注受托企業名稱。產品說明書和標識標簽載明受托企業和受托生產地址。
申請第三類醫療器械產品注冊的,申請人應當向國家藥品監督管理局提交注冊申請資料。
對于科研機構作為注冊申請人申報醫療器械注冊的,可以通過聘用第三方獨立法規事務機構和質量管理體系認證審核機構等,建立質量管理體系,增強質量管理能力,落實醫療器械全生命周期的主體責任。各方應簽訂合同明確注冊人、第三方法規事務機構、質量管理體系認證審核機構、受委托生產企業各自承擔的產品質量責任和質量管理責任。鼓勵建立商業責任保險救濟賠償制度,承擔因上市后醫療器械質量缺陷造成傷害的賠償連帶責任。
(二)生產許可證辦理。注冊人取得醫療器械注冊證后自行生產的,應按照《醫療器械生產監督管理辦法》向省局申辦醫療器械生產許可證,由省局組織現場檢查,符合條件的核發生產許可證。注冊人取得生產許可證后到相應藥品監管部門辦理產品注冊證登記事項變更。
注冊人取得注冊證后不辦理生產許可證,需委托省內其他生產企業生產的,受委托生產企業憑注冊人持有的注冊證和委托合同、質量協議等資料申請辦理醫療器械生產許可證核發或變更,由省局組織現場檢查,符合條件的核發或變更醫療器械生產許可證,并登載受托產品信息。
省內生產企業受托生產省外試點地區注冊人產品的,憑注冊人持有的注冊證和委托合同、質量協議等資料向省局申請醫療器械生產許可證核發或變更,由省局組織現場檢查,符合條件的核發醫療器械生產許可證或辦理醫療器械生產許可證變更。
(三)生產地址登記事項變更辦理。對于注冊人擬通過委托生產方式變更注冊證生產地址的,受托生產企業在我省的,由省局組織開展現場檢查,符合條件的辦理醫療器械生產許可證變更。注冊人提交受托生產企業變更后的醫療器械生產許可證和委托合同,向相應藥品監管部門辦理注冊證登記事項變更。
(四)受托備案。受托生產企業應當向所在地省級藥品監管部門備案,備案時應當提交委托合同、質量協議等資料。
(一)加強組織領導。注冊人制度的試點是改革完善醫療器械審評審批制度和注冊生產管理制度的重大創新,事關醫療器械安全保障水平和產業健康發展,各市市場監管部門和省局有關處室單位應高度重視,全力支持配合開展注冊人制度試點工作。省局成立由分管局領導擔任組長的試點工作組,研究擬定試點工作實施方案和相關管理制度,完善工作機制,加強力量投入,加強職業化、專業化檢查員隊伍建設,扎實推進試點工作開展。
(二)強化監督管理。藥品監管部門要加強對注冊人履行保證醫療器械質量、上市銷售與服務、醫療器械不良事件監測與評價、醫療器械召回等義務情況的監督管理。涉及跨區域試點的,在協調一致的基礎上,確定跨區域監管各方職責劃分,落實日常監管責任主體,確保對醫療器械全生命周期全鏈條監管無縫隙無死角。要建立協同管理、信息共享與結果互認機制。對為醫療器械研制、生產、經營、使用等活動提供產品或者服務的其他相關單位,藥品監管部門可以進行延伸檢查。
(三)鼓勵社會參與。要加強政策宣貫和政策解讀,鼓勵引導相關企業、科研單位積極參與試點實施工作。要加強行業自律,引導注冊人和受托生產企業基于誠信自律的要求開展工作,鼓勵社會力量積極參與企業質量責任保證能力建設,配合做好年度質量管理體系自查、不良事件報告及再評價等工作。
(四)做好評估總結。建立定期問題協商解決機制,對于能在省級層面解決的,由省局協調各方協商解決。須由國家局解決的問題,提出問題解決建議后書面報送國家局,由國家局予以協調解決。試點期間,注重總結提煉實施工作的問題和經驗,主動公開并及時向國家局報送試點工作進展和最新情況。

站點聲明
本網站所提供的信息僅供參考之用,并不代表本網贊同其觀點,也不代表本網對其真實性負責。圖片版權歸原作者所有,如有侵權請聯系我們,我們立刻刪除。如有關于作品內容、版權或其它問題請于作品發表后的30日內與本站聯系,本網將迅速給您回應并做相關處理。
鄭州思途醫療科技有限公司專注于醫療器械產品政策與法規規事務服務,提供產品注冊備案申報代理、臨床試驗、體系建立輔導、分類界定、申請創新辦理服務。
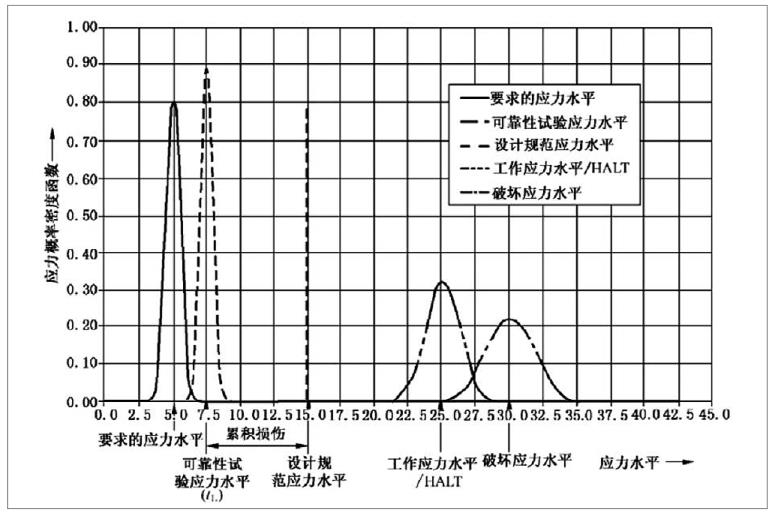
為了確保醫療器械在生命周期內的安全及有效性,注冊人應在設計開發中對其使用期限予以驗證。使用期限的驗證是醫療器械可靠性研究的重要組成部分,制定并驗證合理的使用期限有

國家市場監管總局于官網公布《藥品注冊管理辦法》(以下簡稱“辦法”)已于2020年7月1日起正式施行。國家藥監局從《辦法》修訂的背景、思路、引入了哪些新理念和制度、有哪些鼓

2020年6月29日,國務院發布《化妝品監督管理條例》(國令第727號),規定化妝品新原料需向國務院藥品監督管理部門申請注冊或者備案后才能用于化妝品中。
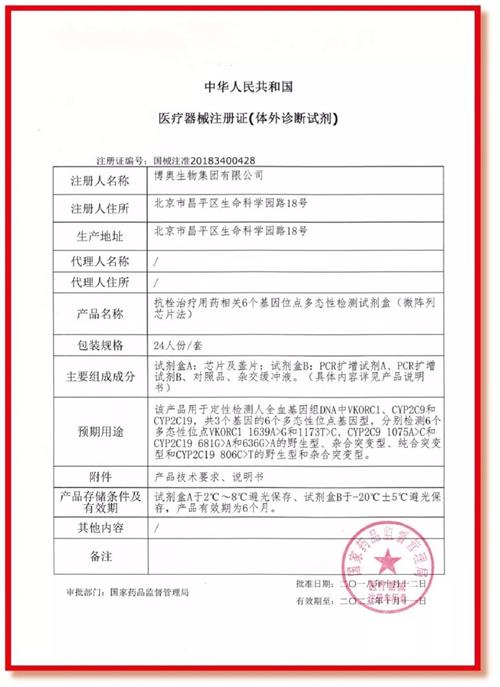
中國是世界上最有潛力的醫療器械市場及最大市場之一,許多醫療器械進出口貿易商或國外有實力的企業希望能夠按照中國藥監局醫療器械注冊監管要求獲得中國市場的上市準入。考慮

醫療器械注冊費是行政性收費,按照注冊單元收取,部分省份不收取醫療器械注冊費用,絕大部分省份還是收取的。本篇文章統計了截止到2022年1月5日各地醫療器械注冊收費標準。

中國能排進世界十大醫療器械制造強國嗎?暫時還不能,因為排進前十的國家都有許多知名械企和世界級醫療器械巨頭;而中國比較出名的可能只有邁瑞了,其他械企仍需努力了。下面

2020年3月17日國家藥監局發布了關于醫療器械主文檔登記事項的公告(2021年第36號),之前給大家介紹過醫療器械主文檔的主要內容、適用的產品注冊類型等信息。今天我們再來向大家介

本文介紹了歐盟醫療器械新法規MDR相比于將替代的MDD法規的幾點新增要求。建議收藏學習。明年5月份起,Medical Devices Regulation(MDR)(2017/745/ EU)將替代原本的Medical Devices Directive (93

潤滑劑類產品在美國根據產品預期用途的不同主要分為人體潤滑劑和患者潤滑劑:其中,人體潤滑劑主要成分為水、丙二醇、羥乙基纖維素、苯甲酸、卡波姆、氫氧化鈉等,作用于生殖

2021年2月5日,湖南省藥監局分別與湖南省計量檢測研究院、湖南新領航檢測技術有限公司、湖南普瑞瑪藥物研究中心有限公司、深圳華通威國際檢驗有限公司、南德認證檢測(中國)有限公
行業資訊
知識分享
八年
醫療器械服務經驗
聯系思途,免費獲得專屬《落地解決方案》及報價
咨詢相關問題或咨詢報價,可以直接與我們聯系
思途CRO——醫療器械注冊臨床第三方平臺
北京市 重慶市 河南省 安徽省 天津市 上海市 河北省 陜西省 山西省 內蒙古 遼寧省 吉林省 黑龍江省 江蘇省 浙江省 福建省 江西省 山東省 湖北省 廣東省 廣西 海南省 四川省 貴州省 云南省

微信公眾號
隨時隨地了解行業動態
客服二維碼
詢價咨詢流程 Copyright ? 2016-2026 思途醫療科技有限公司 版權所有丨備案號:豫ICP備2021016482號-1 ![]() 豫公網安備 41010202003160號 網站地圖
豫公網安備 41010202003160號 網站地圖