




山東省醫療器械工藝用水現場檢查指南(魯藥監械〔2021〕35號)
為規范醫療器械現場檢查,提升檢查員現場檢查能力,指導檢查員對醫療器械生產企業工藝用水控制情況的檢查,提高企業工藝用水質量安全保證水平,依據《醫療器械監督管理條例》


注冊備案 · 臨床試驗 · 體系建立輔導 · 分類界定 · 申請創新
Recommend case

來源:醫療器械注冊代辦 發布日期:2021-12-16 閱讀量:次
附件:包皮切割吻合器注冊審查指導原則(2021年第102號).doc
本指導原則旨在指導注冊申請人對包皮切割吻合器注冊申報的準備及撰寫,同時也為技術審評部門提供參考。
本指導原則是對包皮切割吻合器產品注冊申報資料的一般要求,申請人應依據具體產品特性確定其中內容是否適用,若不適用,需具體闡述理由及相應的科學依據,并依據產品具體特性對注冊申報資料的內容進行充實和細化。
(圖1)")
本指導原則是供注冊申請人和技術審評人員使用的指導性文件,但不包括審評審批所涉及的行政事項,亦不作為法規強制執行,應在遵循相關法規的前提下使用本指導原則。如果有能夠滿足相關法規要求的其他方法,也可以采用,但是需要提供詳細的研究資料和驗證資料。
本指導原則是在現行法規和標準體系以及當前認知水平下制定,隨著法規和標準的不斷完善,以及科學技術的不斷發展,相關內容也將適時進行調整。
本指導原則所涉及的包皮切割吻合器適用于臨床包皮切割縫合手術,為一次性使用。
重復使用的包皮切割吻合器不包含在本指導原則內,可酌情參考本指導原則。
(一)監管信息
1.申請表
1.1產品命名應符合《醫療器械通用名稱命名規則》和國家標準、行業標準中的通用名稱要求,如:包皮切割吻合器,一次性使用包皮切割吻合器、一次性包皮環切吻合器。
1.2根據《醫療器械分類目錄》,分類編碼為02(無源手術器械)-13(手術器械-吻(縫)合器械及材料)-01(吻合器(帶釘))。
1.3注冊單元的劃分應參照《總局關于發布醫療器械注冊單元劃分指導原則的通告》,以產品的技術原理、結構組成、性能指標和適用范圍等因素為劃分依據。
例如:剪刀式和槍式包皮切割吻合器可作為同一注冊單元。
1.4同一注冊單元內,選取典型產品進行檢驗。典型產品是指其檢驗結果能夠涵蓋本注冊單元內全部型號規格的一個或多個產品。
按照“同一注冊單元內,所檢測的產品應當是能夠代表本注冊單元內其他產品安全性和有效性的典型產品”的原則,抽取樣品應能涵蓋該注冊單元全部產品的性能指標。
組成材料、結構、性能等具有差異的產品,建議分別選取典型性型號,進行差異性檢驗。例如:吻合釘材料不同,如鈦與不銹鋼,應分別在選擇同種材料的產品中確定典型產品進行檢驗。
2.產品列表
以表格形式列出包皮切割吻合器的型號、規格、結構及組成、附件,以及每個型號規格的標識(如型號或部件的編號,器械唯一標識等)和描述說明(如尺寸、材質等)。
3.既往溝通記錄
3.1在產品申報前,如果申請人與監管機構針對包皮切割吻合器以會議形式進行了溝通,或者包皮切割吻合器與既往注冊申報相關。應當提供下列內容(如適用):
3.1.1列出監管機構回復的申報前溝通。
3.1.2既往注冊申報產品的受理號。
3.1.3既往申報前溝通的相關資料,如既往申報會議前提交的信息、會議議程、演示幻燈片、最終的會議紀要、會議中待辦事項的回復,以及所有與申請相關的電子郵件。
3.1.4既往申報(如自行撤銷/不予注冊上市申請、臨床試驗審批申請等)中監管機構已明確的相關問題。
3.1.5在申報前溝通中,申請人明確提出的問題,以及監管機構提供的建議。
3.1.6說明在本次申報中如何解決上述問題。
3.2如不適用,應當明確聲明包皮切割吻合器沒有既往申報和/或申報前溝通。
4.主文檔授權信以及其它管理信息等(如適用)
申請人應當對主文檔引用的情況進行說明。申請人應當提交由主文檔所有者或其備案代理機構出具的授權申請人引用主文檔信息的授權信。授權信中應當包括引用主文檔的申請人信息、產品名稱、已備案的主文檔編號、授權引用的主文檔頁碼/章節信息等內容。
(二)綜述資料
1.器械組成
包皮切割吻合器通常由龜頭罩、抵釘座、釘倉蓋、釘倉、手柄、保險扣、調節旋鈕、吻合釘、環形切割刀、推釘片等組成,可配有扎帶等配件。部分產品中包含吻合釘墊圈。產品以無菌形式提供,一次性使用。在結構及組成中至少應明確與人體接觸的組件的材質。
(圖2)")
(圖3)")
2.產品工作原理
利用機械傳動原理,按壓擊發手柄,活動連桿通過推動推釘片推出釘倉內吻合釘,同時帶動環形切割刀,完成切割吻合,從而達到在包皮環切手術中切除過長包皮及吻合的效果。
3.包裝描述
說明所有產品組成的包裝信息。應當說明其無菌屏障系統的信息。說明如何確保最終使用者可清晰地辨識包裝的完整性。
4.研發歷程
闡述申請包皮切割吻合器的研發背景和目的。如有參考的同類產品或前代產品,應當提供同類產品或前代產品的信息,并說明選擇其作為研發參考的原因。
5.適用范圍
適用于臨床包皮的切割縫合手術。
6.預期使用環境
預期使用環境為醫療機構。
7.禁忌證
(1)急性尿路感染、手術部位急性炎癥(如包皮及龜頭感染等)、水腫期患者禁用;
(2)隱匿性陰莖者禁用;
(3)患有系統性疾病(如出血傾向、低蛋白血癥、嚴重心血管疾病等)及精神疾病等禁忌手術者禁用;
(4)糖尿病患者在血糖未控制至正常范圍之前禁用;
(5)陰莖癌患者禁用;
(6)近期使用過有抗凝血功能藥物者禁用;
(7)對產品使用的原材料過敏者禁用。
8.產品的不良事件歷史記錄
根據文獻記載及相關生產企業不良事件信息分析,包皮切割吻合器產品在臨床中出現的可疑不良事件主要有:無法有效切斷組織,無法正常吻合,落釘漏釘,手柄斷裂,保險扣滑落,組件無法預期使用,使用完畢產品無法退出,無菌包裝破損,吻合釘在規定的時間內無法自行脫落等。申請人應當收集并全面分析上報包皮切割吻合器相關可疑不良事件,以促進該類產品的進一步技術更新,最大限度地控制醫療器械潛在風險,保證該類產品安全有效地使用。申請人在風險分析時應關注同品種產品的不良事件歷史記錄。
(三)非臨床資料
1.產品風險管理資料
申請人應建立風險管理控制程序,并對包皮切割吻合器產品的設計開發、原材料采購、生產加工、包裝、滅菌、運輸、貯存、使用等產品生命周期全過程實施風險管理(按照YY/T 0316《醫療器械 風險管理對醫療器械的應用》標準的要求)。申請人在產品注冊上市前,應對風險管理過程進行評審。應建立風險管理文檔,風險管理文檔應至少包括以下信息:
1.1可能影響產品安全性的特征問題清單。應判定醫療器械與安全性有關特征的問題,但識別風險的來源并不局限于此(參考YY/T 0316附錄C)。
1.2產品有關的已知和可預見危害的清單。危險(源)分析是否全面,申請人應編寫在正常使用和非正常使用兩種條件下,與產品有關的已知的和可預見的危險(源)文件(參考YY/T 0316附錄E)。已識別的風險應至少包括但不局限于以下方面:
(1)設計和開發不當產生的危害。
(2)原材料的生物學和化學危害:材料或材料來源變化;材料的生物相容性和對生殖器官的影響。
(3)生產制造可能產生的危害:污染;微粒殘留;添加劑、粘接劑、助劑、輔劑的殘留;工藝用水;生產環境潔凈度;滅菌方式對產品不適宜,滅菌不完全,環氧乙烷殘留量不合格等。
(4)不正確使用產生的危害:產品選擇與使用不當;未按照說明書中操作方法操作;重復使用等。
(5)運輸和貯藏產生的危害:包裝破損;貯藏環境不當;標識不清等。
1.3風險評價、風險控制措施以及剩余風險評價匯總表。申請人應在產品生命全周期中對風險進行管理控制,以使剩余風險在可接受范圍內。
表2示例性地給出了包皮切割吻合器產品可能涉及的部分危險、可預見的事件序列、危險情況和可能發生的傷害之間的關系(根據YY/T 0316附錄E)。申請人還應結合自身產品特點確定其他危險,并針對各項危險,采取控制措施,確保危險降低到可接受的程度。
表2 部分危險、可預見的事件序列、危險情況和可發生的傷害之間的關系
| 危險 | 可預見的事件序列 | 危險情況 | 傷害 |
| 設計開發危險 | 產品機械系統設計不當;器身及吻合釘材料選擇不當,導致不能有效切割或吻合 | 使用過程中損傷生殖器官或無法使用 | 造成損傷、感染或額外傷害 |
| 生物學和化學危險 | 選用不適當的材料;滅菌未確認或未按已確認的參數滅菌;超過有效期使用;包裝不符合要求或老化;零部件生銹;未按要求對生產環境進行控制;零部件未按要求清洗;清洗用水不符合要求;使用完后,未按醫療垃圾處理 | 生物相容性不符合要求;被污染的產品與人體接觸;有菌或有毒物質影響環境 | 產生刺激、過敏、感染等危害;造成環境污染或交叉感染 |
| 制造過程危險 | 采購不當;零部件加工精度不當,裝配調試不當;不合格品未被檢出;發生缺釘/掉釘現象;包裝不當;滅菌有效性未被充分確認/驗證 | 無法正常使用產品;性能不合格的產品使用于人體 | 手術中斷,延長手術時間;手術失敗或對患者造成機械損傷 |
| 運輸和貯藏危險 | 不恰當的包裝導致污染;貯藏環境不當;未能按運輸儲存要求對產品進行防護,造成產品破損、污染;防護不當導致運輸中吻合釘脫位/脫落 | 被污染的產品與人體接觸;使用過程中無法正常吻合 | 造成感染;造成損傷、出血 |
| 使用危險 | 型號規格選擇不當,造成錯誤使用;未明示應由經培訓的專業人員使用;不完整的使用說明書,造成操作錯誤;性能特征不恰當的描述、不適當的使用規范,造成錯誤使用;非預期使用;手柄斷裂;使用者未按規范程序使用;重復使用 | 規格不當;使用過程中損傷生殖器官或無法使用;產品重復使用 | 造成損傷、感染或額外傷害;交叉感染;手術失敗 |
2.產品技術要求
根據《醫療器械注冊與備案管理辦法》的要求,產品技術要求應符合國家標準、行業標準和有關法律、法規的要求。在此基礎上,申請人應根據產品的特點制定保證產品安全有效、質量可控的技術要求。產品技術要求及試驗方法均應經過驗證。常見的技術指標包括但不限于:
2.1外觀及尺寸
2.1.1產品外表面應平整、光滑,無鋒棱、毛刺及裂紋。器身上如有標志或刻度,應清晰。
2.1.2環形切割刀切口直徑。
2.1.3吻合釘數量、尺寸。
2.2物理性能
2.2.1硬度:由申請人根據不同材料特性及設計需求,制定抵釘座、環形切割刀的硬度要求。
2.2.2表面粗糙度:與患者接觸的產品金屬組件外露表面粗糙度Ra應不大于0.8 μm。
2.2.3環形切割刀鋒利度:環形切割刀刃口應鋒利,不得有卷刃、崩刃,當切割3—0真絲捻制不涂層縫合線時,其切割力應不大于1.6 N。
2.3化學性能
2.3.1耐腐蝕性能:與患者接觸的產品金屬組件的耐腐蝕性能(參考YY/T 0149中5.4 b)的規定)。
2.3.2由申請人根據不同材料特性決定是否對產品中組件、配件的化學性能(重金屬、酸堿度、紫外吸光度等)提出要求。若滅菌使用的方法容易產生殘留,應明確殘留物的名稱及殘留量要求。
2.4使用性能
2.4.1產品的需裝配組件(龜頭罩等)與器身架應能順利地裝配和拆卸;各移動部位應能順利推動,不得有卡阻、松動現象。
2.4.2產品的保險扣開閉應靈活,使用應安全;彈簧應有足夠的彈性,松開手柄時能迅速復位。
2.4.3產品應具有良好的吻合和切割性能,吻合后的吻合釘應成規定的形狀。
2.5無菌
3.研究資料
根據所申報的產品,提供適用的研究資料。
3.1產品性能研究
應當提供產品性能研究資料以及產品技術要求的研究和編制說明,包括功能性、安全性指標(如化學性能、物理性能、微生物性能)以及與質量控制相關的其他指標的確定依據。產品的性能要求及試驗方法可參考相應國家標準、行業標準的的適用部分進行制定(如YY/T 0245《吻(縫)合器 通用技術條件》等)。
3.2生物相容性評價研究
對產品中與患者短期接觸的組件,如抵釘座、釘倉等應按照GB/T 16886《醫療器械生物學評價》系列標準進行生物相容性評價。一般應評價的項目包括細胞毒性、致敏和皮內反應等。
對產品中與患者長期接觸的組件,如吻合釘、吻合釘墊圈等應按照GB/T 16886《醫療器械生物學評價》系列標準進行生物相容性評價。一般應評價的項目包括細胞毒性、致敏、皮內反應等。
吻合釘目前多采用鈦、鈦合金或純鉭、不銹鋼材料。吻合釘一般應選用符合GB/T 13810中鈦或鈦合金材料化學成分要求的純鈦、鈦合金材料,或符合YY/T 0245、ISO 13782中純鉭材料化學成分要求的純鉭材料,或符合GB 4234.1中不銹鋼材料化學成分要求的不銹鋼材料。對于采用以上材料制作的吻合釘可豁免生物學試驗,并將吻合釘成分證明資料作為生物相容性評價研究資料的一部分。選用其他材料(如其他不銹鋼材料、其他金屬材料)或表面改性處理的純鈦、鈦合金、純鉭等制成的吻合釘,應按照GB/T 16886《醫療器械生物學評價》系列標準對吻合釘進行生物相容性評價研究,一般包括但不限于細胞毒性、致敏、皮內反應、急性毒性、亞慢性毒性、遺傳毒性和植入后局部反應。
部分產品結構組成中包括吻合釘墊圈,應按照GB/T 16886《醫療器械生物學評價》系列標準對吻合釘墊圈進行生物相容性評價研究,一般應評價的項目包括細胞毒性、致敏、皮內反應。
生物相容性評價研究資料應當包括:生物相容性評價的依據和方法;產品所用材料的描述及與人體接觸的性質;實施或豁免生物學試驗的理由和論證;對于現有數據或試驗結果的評價。
3.3滅菌工藝研究
應明確滅菌工藝(方法和參數)及其選擇依據和無菌保證水平(SAL),并提供滅菌確認報告,無菌保證水平(SAL)應保證達到1×10-6。滅菌過程的選擇應至少考慮以下因素:產品與滅菌過程的適應性;包裝材料與滅菌過程的適應性;滅菌對產品安全有效性的影響。
若滅菌使用的方法易出現殘留,應明確殘留物的名稱、限量及其確定依據、采取的處理措施及相應的殘留量檢測報告。
3.4產品有效期和包裝研究
3.4.1有效期研究資料
貨架有效期包括產品有效期和包裝有效期。產品有效期驗證可采用加速老化或實時老化的研究,加速老化試驗研究的具體要求可參考YY/T 0681.1,應遵循極限試驗等原則。
對于包裝的有效期驗證,建議申請人提交在選擇恰當的材料和包裝結構合格后的最終成品包裝的初始完整性和維持完整性的檢測結果。在進行加速老化試驗研究時應注意產品選擇的環境條件的老化機制應與在實時正常使用環境老化條件下真實發生產品老化的機制相匹配。
3.4.2包裝研究資料
產品包裝驗證可依據有關標準進行(如GB/T 19633、YY/T 0681系列標準等),提交產品的包裝驗證報告。包裝材料的選擇應至少考慮以下因素:包裝材料的物理化學性能;包裝材料與產品的適應性;包裝材料與成型和密封過程的適應性;包裝材料與滅菌過程的適應性;包裝材料所能提供的物理、化學和微生物屏障保護;包裝材料與使用者使用時的要求(如無菌開啟)的適應性;包裝材料與標簽系統的適應性;包裝材料與貯存運輸過程的適應性。
3.5其他資料
證明產品安全性、有效性的其他研究資料。
(四)臨床評價資料
包皮切割吻合器列入《免于臨床評價醫療器械目錄》,申請人無需提交臨床評價資料。
對于與《免于臨床評價醫療器械目錄》描述不相符的一次性使用包皮切割吻合器產品,例如新型作用機理產品,應按照《醫療器械臨床評價技術指導原則》第六條或第七條的要求提交臨床評價資料。
(五)產品說明書和標簽樣稿
產品說明書、標簽應符合《醫療器械說明書和標簽管理規定》的要求,并可參考YY/T 0466.1《醫療器械 用于醫療器械標簽、標記和提供信息的符號》等相關標準。所提交的文本和標簽樣稿應內容清晰、完整。說明書中的適用范圍、禁忌證、注意事項、警示信息、有效期等信息應與產品綜述資料、研究資料和臨床評價資料中所描述及驗證的內容一致。產品說明書中聲稱的產品結構組成、尺寸和其他技術信息應與產品技術要求、檢驗報告中內容一致。產品說明書中應注明產品的交付狀態,明確包裝材料、包裝形式、使用說明、注意事項、廢棄物處置。
同時,在說明書的注意事項或警示信息中應至少包含以下內容:
1.本產品的使用必須符合醫療機構相關操作規范及法規要求,僅限于經培訓的醫務人員使用;
2.產品為一次性使用,禁止重復使用;
3.應提示滅菌方式,包裝破損禁止使用;超出有效期時禁止使用;
4.應選用型號規格與陰莖大小相近的產品進行手術;
5.在未完整閱讀使用說明書之前請勿嘗試操作該產品,任何的不謹慎操作都將給手術帶來風險;
6.產品使用后應按照醫療機構的衛生管理規范進行處置;接觸過體液的產品應特別處置以防生物污染的發生;
7.包皮粘接、系帶過短及其他先天異常者,應先進行處理并止血等,之后再使用本產品;
8.使用前應檢查外觀和保險扣是否正常,有異常時禁止使用;直到產品準備好要擊發,方可釋放保險扣;
9.要一次擊發到底,切勿部分擊發器械,以確保吻合釘正確成型及包皮正確切割;
10.手術過程中出現包皮沒有完全切斷時,可用手術剪加以修剪;罕見的明顯出血經按壓無效時可進行縫合止血,不得進行重復切割吻合操作;術后局麻處和術口部位少許淤腫屬正常現象,會自行消退;
11.患者術后應返院檢查;吻合釘在規定的時間內如未脫落應返院進行處理。
(六)質量管理體系文件
1.申請人基本情況表。若申報產品有多個研制、生產場地,應概述每個研制、生產場地的實際情況。
2.申請人組織機構圖。
3.生產企業總平面布置圖、生產區域分布圖。
4.生產過程有凈化要求,應當提供有資質的檢測機構出具的環境檢測報告(附平面布局圖)復印件。
5.應列明產品生產過程中所需原材料的名稱、牌號、供應商、符合標準等基本信息。主要原材料應具有穩定的供貨渠道以保證產品質量,需提供原材料供應商的資質證明文件及質量協議,需明確所用原材料(包括初包裝材料)的質量控制要求。應重點關注吻合釘原材料的質量控制措施。
6.應提交產品的生產工藝管理控制文件,詳細說明產品的生產工藝和步驟,列出工藝圖表,闡述生產工藝過程的質量控制標準及控制措施。生產工藝應已通過驗證,能夠生產出質量穩定、安全有效的產品,在注冊質量管理體系核查中,對此項內容進行核查。
應注明特殊過程和關鍵工序(如:裝配、焊接、清洗、內包封口、滅菌等),明確其過程控制點及控制參數,對生產工藝的可控性、穩定性應進行確認。
吻合釘若選用表面改性處理(涂層、酸蝕等)的金屬材料(如純鈦、鈦合金材料、純鉭、不銹鋼等),需要給出改性層或涂層的元素成分、組織結構、理化性能、結合強度等信息及其相關的制備工藝。
對生產加工過程使用的所有輔劑、助劑等添加劑均應說明使用劑量、對殘留量的控制措施和接受標準,并提供檢驗報告和安全性評價報告。
7.主要生產設備和檢驗設備(包括進貨檢驗、過程檢驗、出廠最終檢驗所需的相關設備、環境監測設備)目錄。
8.質量管理體系自查報告。
9.應當提供擬核查產品與既往已通過核查產品在生產條件、生產工藝等方面的對比說明(如適用)。
[1]GB/T 12672,丙烯腈-丁二烯-苯乙烯(ABS)樹脂[S].
[2]GB/T 1220,不銹鋼棒[S].
[3]GB/T 13810,外科植入物用鈦及鈦合金加工材料[S].
[4]GB/T 230.1, 金屬材料 洛氏硬度試驗方法 第1部分:試驗方法[S].
[5]GB/T 3280, 不銹鋼冷軋鋼板和鋼帶[S].
[6]GB 4234.1, 外科植入物 金屬材料 第1部分:鍛造不銹鋼[S].
[7]GB/T 4240, 不銹鋼絲[S].
[8]GB/T 4340.1, 金屬材料 維氏硬度試驗 第1部分:試驗方法[S].
[9] GB/T 14233,系列標準,醫用輸液、輸血、注射器具檢驗方法[S].
[10] GB/T 16886,系列標準,醫療器械生物學評價[S].
[11] GB 18279,系列標準,醫療保健產品滅菌 環氧乙烷[S].
[12] GB 18280,系列標準,醫療保健產品滅菌 輻射[S].
[13] GB/T 19633,系列標準, 最終滅菌醫療器械包裝[S].
[14] YY/T 0149, 不銹鋼醫用器械 耐腐蝕性能試驗方法[S].
[15] YY/T 0171, 外科器械 包裝、標志和使用說明書[S].
[16] YY/T 0245, 吻(縫)合器通用技術條件[S].
[17] YY 0334, 硅橡膠外科植入物通用要求[S].
[18] YY/T 0466.1, 醫療器械用于醫療器械標簽、標記和提供信息的符號第1部分:通用要求[S].
[19] YY/T 0681系列標準, 無菌醫療器械包裝試驗方法[S].
[20] ISO 13782, Implants for surgery - Metallic materials - Unalloyed tantalum for surgical implant applications/外科植入物-金屬材料-外科植入用純鉭材料[S].
[21] 中華人民共和國藥典(2020版)
湖北省藥品監督管理局審評中心

站點聲明
本網站所提供的信息僅供參考之用,并不代表本網贊同其觀點,也不代表本網對其真實性負責。圖片版權歸原作者所有,如有侵權請聯系我們,我們立刻刪除。如有關于作品內容、版權或其它問題請于作品發表后的30日內與本站聯系,本網將迅速給您回應并做相關處理。
鄭州思途醫療科技有限公司專注于醫療器械產品政策與法規規事務服務,提供產品注冊備案申報代理、臨床試驗、體系建立輔導、分類界定、申請創新辦理服務。
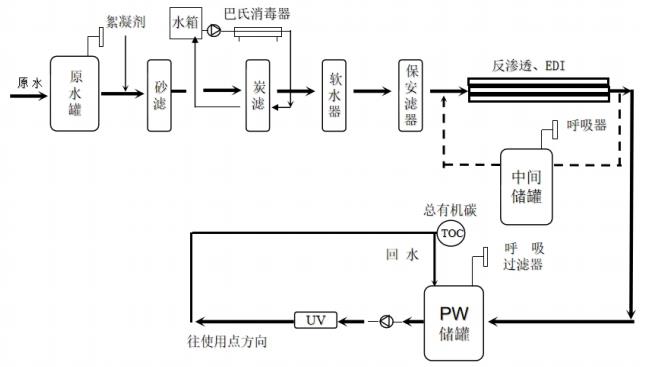
為規范醫療器械現場檢查,提升檢查員現場檢查能力,指導檢查員對醫療器械生產企業工藝用水控制情況的檢查,提高企業工藝用水質量安全保證水平,依據《醫療器械監督管理條例》

為深入貫徹落實中共中央辦公廳、國務院辦公廳印發的《關于深化審評審批制度改革鼓勵藥品醫療器械創新的意見》(廳字〔2017〕42號),按照國家藥品監督管理局《關于擴大醫療器械
為規范醫療器械注冊人跨區域委托生產的監督管理,推進長江三角洲區域醫療器械跨區域監管,根據《中共中央辦公廳國務院辦公廳關于深化審評審批制度改革鼓勵藥品醫療器械創新的意見》《

附件:血液透析用水處理設備注冊審查指導原則(2024年修訂版)(2024年第19號).doc血液透析用水處理設備注冊審查指導原則(2024年修訂版)本指導原則旨在指導注冊申請人對血液透析用水處理設備注冊申報資料的準備及撰寫,同時也為技術審評部門提供參考。本指導原則是對血液透析用水處理設備的一般要求,注冊申請人依據產品的具體特性確定其中內容是否適用。若不適用,要具體闡述理由及相應的科學依據,并依據產

本指導原則旨在幫助和指導注冊申請人對一次性使用腹部穿刺器注冊申報資料進行準備,以滿足技術審評的基本要求。同時有助于審評機構對該類產品進行科學規范的審評,提高審評工

國家藥監局關于發布免于臨床試驗的體外診斷試劑臨床評價技術指導原則的通告(2021年第74號)發布時間:2021-09-24為指導體外診斷試劑的臨床評價工作,根據

國家藥監局藥審中心關于發布《抗腫瘤藥物臨床試驗中SUSAR分析與處理技術指導原則》的通告(2024年第42號) 發布日期:20241010

國家市場監督管理總局令 第47號 《醫療器械注冊與備案管理辦法》已經2021年7月22日市場監管總局第11次局務會議通過,現予公布,自2021年10月1日起施行。 附件: 醫療器械注冊與備案管

國家藥監局器審中心關于發布影像型超聲診斷設備(第三類)注冊審查指導原則(2023年修訂版)的通告(2024年第29號)發布時間:2024-10-14為進一步規范

附件:醫用透明質酸鈉創面敷料注冊審查指導原則(2024年第21號).doc醫用透明質酸鈉創面敷料注冊審查指導原則本指導原則旨在指導注冊申請人準備及撰寫醫用透明質酸鈉創面敷料注冊申報資料,同時也為技術審評部門審查注冊申報資料提供參考。本指導原則是對醫用透明質酸鈉創面敷料產品的一般要求,注冊申請人應依據產品的具體特性確定其中內容是否適用,若不適用,需具體闡述理由及相應的科學依據,并依據產品的具體特性對
行業資訊
知識分享
八年
醫療器械服務經驗
聯系思途,免費獲得專屬《落地解決方案》及報價
咨詢相關問題或咨詢報價,可以直接與我們聯系
思途CRO——醫療器械注冊臨床第三方平臺
北京市 重慶市 河南省 安徽省 天津市 上海市 河北省 陜西省 山西省 內蒙古 遼寧省 吉林省 黑龍江省 江蘇省 浙江省 福建省 江西省 山東省 湖北省 廣東省 廣西 海南省 四川省 貴州省 云南省

微信公眾號
隨時隨地了解行業動態
客服二維碼
詢價咨詢流程 Copyright ? 2016-2026 思途醫療科技有限公司 版權所有丨備案號:豫ICP備2021016482號-1 ![]() 豫公網安備 41010202003160號 網站地圖
豫公網安備 41010202003160號 網站地圖