淺談病例報(bào)告表設(shè)計(jì)/填寫與更正(附病例報(bào)告表模板)
病例報(bào)告表(CRF)是臨床試驗(yàn)中記錄臨床資料的表格,每一受試者有關(guān)試驗(yàn)的資料均應(yīng)記錄在預(yù)先按試驗(yàn)方案設(shè)計(jì)的病例報(bào)告表中。它們依據(jù)原始記錄而填寫,以便申辦者對(duì)不同試驗(yàn)單


注冊(cè)備案 · 臨床試驗(yàn) · 體系建立輔導(dǎo) · 分類界定 · 申請(qǐng)創(chuàng)新
Recommend case

遠(yuǎn)紅外穴位貼/前列腺遠(yuǎn)紅外穴位貼臨床試驗(yàn)注冊(cè)案例
醫(yī)用疤痕/遠(yuǎn)紅外治療凝膠臨床試驗(yàn)注冊(cè)案例
脈沖磁場(chǎng)穴位治療儀同品種比對(duì)產(chǎn)品注冊(cè)案例

生物玻璃創(chuàng)面無(wú)機(jī)敷料醫(yī)療器械產(chǎn)品注冊(cè)證申辦案例

牙齒脫敏劑醫(yī)療器械注冊(cè)免臨床GMP案例

醫(yī)用冷凍箱醫(yī)療器械注冊(cè)免臨床體系建立案例

醫(yī)用冷藏箱醫(yī)療器械注冊(cè)GMP體系認(rèn)證案例
定制式活動(dòng)義齒醫(yī)療器械注冊(cè)證變更案例

業(yè)務(wù)咨詢:186-0382-3910(高先生,微信同)
加急電話:186-0382-3910
在線客服:周一至周日8:00-22:00
來(lái)源:醫(yī)療器械注冊(cè)代辦 發(fā)布日期:2023-10-10 閱讀量:次
?
兒童受試者屬于弱勢(shì)群體(vulnerable subjects),參加臨床試驗(yàn)時(shí),應(yīng)給予更多的保護(hù)和尊重。前兩天分析了臨床項(xiàng)目管理中的進(jìn)度管理,今天和大家深度討論兒童受試者知情同意的年齡問(wèn)題和知情權(quán)的保護(hù)。主要分為以下兩個(gè)層面:年齡界限的討論、年齡分段知情的管理。
")
小編通過(guò)文獻(xiàn)查閱和各種渠道的資源,了解到除了本文以下章節(jié)中提到的兩個(gè)法規(guī)和指導(dǎo)原則以外,在ICH-GCP, 中國(guó)GCP,以及其他法規(guī)或指南性文件中,均沒有提到關(guān)于兒童知情同意年齡的相關(guān)信息,僅僅是提到保護(hù)兒童受試者,對(duì)兒童受試者的知情同意過(guò)程需法定監(jiān)護(hù)人參加。
2016年3月1日國(guó)家局發(fā)布的《兒科人群藥物臨床試驗(yàn)技術(shù)指導(dǎo)原則》(以下簡(jiǎn)稱“指導(dǎo)原則”)中,2.2.2章節(jié)中提到獲取兒童知情同意書年齡的要求。
(1)在我國(guó),通常10周歲以上(含10周歲)的兒科人群應(yīng)參與知情同意并簽署知情同意書。
(2)對(duì)于一些特殊疾病,如智力認(rèn)知發(fā)育障礙,能否參與或簽署知情同意取決于能力而不僅是年齡。
然而,該指導(dǎo)原則中關(guān)于兒童受試者簽署知情同意書的年齡界限的依據(jù)是來(lái)源于我國(guó)民法總則,民法總則在該指導(dǎo)原則發(fā)布1年后,就做了更新,其中更新的內(nèi)容涉及到對(duì)于兒童年齡界限調(diào)整。讓我們一起來(lái)看一下,2017版民法總則的相關(guān)內(nèi)容。
中華人民共和國(guó)民法總則(2017年3月15日發(fā)布,以下簡(jiǎn)稱“民法總則”)明確了未成年人的年齡界限:
(1)十八周歲以上的自然人為成年人。不滿十八周歲的自然人為未成年人(第十七條)
(2)八周歲以上的未成年人為限制民事行為能力人……(第十九條)
(3)不滿八周歲的未成年人為無(wú)民事行為能力人……(第二十條)
因此,兒童受試者簽署知情同意的年齡應(yīng)由“指導(dǎo)原則”中規(guī)定的10周歲下調(diào)至8周歲,更加體現(xiàn)出對(duì)于兒童受試者知情權(quán)的保護(hù)和尊重。同時(shí),這也是與兒童生長(zhǎng)發(fā)育和心理成熟度,以及社會(huì)認(rèn)知的不斷發(fā)展和變化相適應(yīng)。
")
9周歲兒童受試者臨床
目前行業(yè)上,兒童受試者的知情同意書的執(zhí)行現(xiàn)狀有兩種情況:“一刀切”和“分段”管理。
“一刀切”是指臨床試驗(yàn)中只有一個(gè)版本的兒童受試者知情同意書。無(wú)論兒童受試者的年齡如何,只要達(dá)到簽署知情同意書的界限,就統(tǒng)一簽署一個(gè)版本的知情同意書。大多數(shù)國(guó)內(nèi)的項(xiàng)目都是這樣的實(shí)際操作,因?yàn)榉ㄒ?guī)沒有明確對(duì)于按照年齡區(qū)分知情同意的告知內(nèi)容而做詳細(xì)要求。
“分段管理”是指臨床試驗(yàn)中有不同版本的知情同意書,內(nèi)容設(shè)計(jì)是根據(jù)達(dá)到簽署知情同意書的兒童受試者年齡的認(rèn)知情況而劃分為2個(gè)以上不同的版本,例如8-10歲,11-12歲,13-14歲等。很多外資企業(yè)申辦的臨床試驗(yàn)會(huì)執(zhí)行分段管理,充分尊重兒童受試者的知情同意權(quán)。
補(bǔ)充:
受試者為兒童時(shí)如何簽署知情同意書?請(qǐng)看兒童臨床試驗(yàn)知情同意及倫理考量
兒童知情同意書簽署幾份?答:簽署兩份,醫(yī)院保留一份,受試者一份。
咨詢")
站點(diǎn)聲明
本網(wǎng)站所提供的信息僅供參考之用,并不代表本網(wǎng)贊同其觀點(diǎn),也不代表本網(wǎng)對(duì)其真實(shí)性負(fù)責(zé)。圖片版權(quán)歸原作者所有,如有侵權(quán)請(qǐng)聯(lián)系我們,我們立刻刪除。如有關(guān)于作品內(nèi)容、版權(quán)或其它問(wèn)題請(qǐng)于作品發(fā)表后的30日內(nèi)與本站聯(lián)系,本網(wǎng)將迅速給您回應(yīng)并做相關(guān)處理。
鄭州思途醫(yī)療科技有限公司專注于醫(yī)療器械產(chǎn)品政策與法規(guī)規(guī)事務(wù)服務(wù),提供產(chǎn)品注冊(cè)備案申報(bào)代理、臨床試驗(yàn)、體系建立輔導(dǎo)、分類界定、申請(qǐng)創(chuàng)新辦理服務(wù)。

病例報(bào)告表(CRF)是臨床試驗(yàn)中記錄臨床資料的表格,每一受試者有關(guān)試驗(yàn)的資料均應(yīng)記錄在預(yù)先按試驗(yàn)方案設(shè)計(jì)的病例報(bào)告表中。它們依據(jù)原始記錄而填寫,以便申辦者對(duì)不同試驗(yàn)單
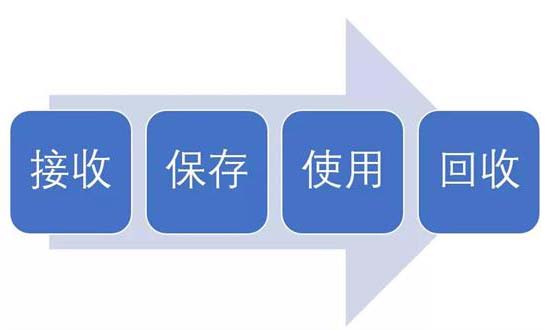
試驗(yàn)用藥品是指用于臨床試驗(yàn)的試驗(yàn)藥物、對(duì)照藥品。試驗(yàn)用藥品滲透到了臨床試驗(yàn)過(guò)程中的每一個(gè)步驟,包括藥物的生產(chǎn)、包裝、運(yùn)輸、保存、使用、回收等。今天我們從臨床試驗(yàn)中

臨床研究協(xié)調(diào)員(全稱Clinical Research Coordinator,簡(jiǎn)稱CRC):指經(jīng)主要研究者授權(quán),在臨床試驗(yàn)中協(xié)助研究者進(jìn)行非醫(yī)學(xué)性的相關(guān)事務(wù)性工作,是臨床試驗(yàn)的參與者。 臨床試驗(yàn)現(xiàn)場(chǎng)管理組

俗話說(shuō)“知己知彼,百戰(zhàn)不殆”,對(duì)于作為CRC的我們,自認(rèn)為對(duì)CRA其實(shí)已經(jīng)很了解了,但是在我們工作過(guò)程有一個(gè)角色平時(shí)接觸不到,但是卻又繞不開躲不過(guò),尤其是面對(duì)滿屏EDC query的

生物樣品分析在臨床試驗(yàn)中起著非常重要的作用,是臨床試驗(yàn)研究中一個(gè)關(guān)鍵的環(huán)節(jié),在臨床試驗(yàn)過(guò)程中,CRC很多時(shí)候也會(huì)被授權(quán)參與生物樣本的管理,因此我們小伙伴們也要掌握這項(xiàng)

回顧以往的文章,自己還真的沒有寫關(guān)于CRC管理方面的心得,今天就六個(gè)方面淺談一下個(gè)人的心得體會(huì)。

方案偏離和方案違背是我們?cè)谄綍r(shí)工作過(guò)程中,非常容易遇到的一種情況,今天,我們就來(lái)聊一下應(yīng)該 如何做才能更好的降低方案偏離/違背發(fā)生呢? 1.定義 其實(shí)GCP中并未對(duì)方案偏離

大家在項(xiàng)目中,是否遇到過(guò)ICF更新的情況?在ICF更新后,已由受試者簽署的知情同意書,是否需要重新簽署呢?又有哪些情形不需要重新簽署呢?本期我們繼續(xù)跟大家分享知情同意書實(shí)

當(dāng)今,隨著新的GCP法規(guī)的全面實(shí)施,并伴隨著各項(xiàng)監(jiān)督抽查在全國(guó)范圍內(nèi)陸續(xù)開展,醫(yī)療器械臨床試驗(yàn)的各種規(guī)定越來(lái)越嚴(yán)格了。除了過(guò)程中試驗(yàn)的質(zhì)量必須得到有效的控制外,整個(gè)試

臨床數(shù)據(jù)管理員(DM)能力和要求?數(shù)據(jù)管理員在整個(gè)臨床研究中要密切注意研究中的關(guān)鍵變量的數(shù)據(jù)質(zhì)量,確保它們百分之百準(zhǔn)確無(wú)誤。此外,臨床研究方案還應(yīng)該對(duì)試驗(yàn)的質(zhì)量控
行業(yè)資訊
?
?
?
?
?
?
知識(shí)分享
?
?
?
?
?
?
法規(guī)文件
?
?
?
?
?
?
八年
醫(yī)療器械服務(wù)經(jīng)驗(yàn)
聯(lián)系思途,免費(fèi)獲得專屬《落地解決方案》及報(bào)價(jià)
咨詢相關(guān)問(wèn)題或咨詢報(bào)價(jià),可以直接與我們聯(lián)系
思途CRO——醫(yī)療器械注冊(cè)臨床第三方平臺(tái)
北京市 重慶市 河南省 安徽省 天津市 上海市 河北省 陜西省 山西省 內(nèi)蒙古 遼寧省 吉林省 黑龍江省 江蘇省 浙江省 福建省 江西省 山東省 湖北省 廣東省 廣西 海南省 四川省 貴州省 云南省

微信公眾號(hào)
隨時(shí)隨地了解行業(yè)動(dòng)態(tài)
客服二維碼
詢價(jià)咨詢流程 Copyright ? 2016-2026 思途醫(yī)療科技有限公司 版權(quán)所有丨備案號(hào):豫ICP備2021016482號(hào)-1 ![]() 豫公網(wǎng)安備 41010202003160號(hào) 網(wǎng)站地圖
豫公網(wǎng)安備 41010202003160號(hào) 網(wǎng)站地圖